
Resistansi Acyclovir pada Infeksi Virus Varicella Zoster
- 24 Jul 2025
- 23 menit membaca

Resistansi Acyclovir pada Infeksi Virus Varicella Zoster
Annisa Fildza Hashfi, Nurrachmat Mulianto
Bagian/KSM Ilmu Kesehatan Kulit dan Kelamin
Fakultas Kedokteran Universitas Sebelas Maret/ RSUD dr. Moewardi Surakarta
Pendahuluan
Varicella zoster virus (VZV) juga disebut dengan human herpesvirus 3 (HHV-3, HHH) merupakan golongan herpervirus dengan susunan DNA untaian ganda.[1] Virus ini secara alami menginfeksi manusia tanpa melalui reservoir hewan. Target utamanya adalah limfosit T, sel epitel dan ganglia. Infeksi primer menyebabkan varisela yang menjadi laten di saraf ganglion. Virus laten yang aktif kembali menyebabkan herpes zoster yang dapat diperberat dengan postherpetic neuralgia (PHN). Komplikasi serius dapat terjadi akibat infeksi virus ini berupa meningoencephalitis, myelitis, kelumpuhan saraf kranial, vasculopathy, keratitis, retinopati, ulser, hepatitis, dan pankreatitis.[2,3,4]
Insidensi varisela di dunia dilaporkan berkisar dari 13 sampai 16 kasus per 1000 orang per tahun.[2] Angka kejadian varisela di Indonesia belum diketahui secara pasti, namun dalam studi oleh Sondakh dkk pada tahun 2012 di Poliklinik Kulit dan Kelamin RS Prof. Dr. RD. Kandou Manado menunjukkan terdapat 27 kasus varisela yang ditemukan dalam satu tahun, sedangkan berdasarkan data dari poliklinik umum Ilmu Kesehatan Anak Rumah Sakit Cipto Mangunkusumo Jakarta pada tahun 2005-2010 tercatat 77 kasus varisela tanpa komplikasi.[5] Data insidensi herpes zoster pada 13 rumah sakit pendidikan di Indonesia cukup bervariasi dan diperkirakan mencapai sekitar 2232 pasien pada tahun 2011-2013. Puncak kasus herpes zoster terjadi pada usia 45-64 tahun yang merupakan 37,95% dari total kasus herpes zoster. Kelompok usia yang sama juga mengalami puncak kasus terjadinya PHN yakni mencapai 42% dari total kasus PHN pada pasien dengan herpes zoster.[6] Catatan medis di 4 benua dunia menunjukkan insidensi herpes zoster mencapai 8-12 kasus per 1000 orang tiap tahun. Terdapat setidaknya 1,5 juta kasus herpes zoster baru per tahun.[4]
Acyclovir merupakan analog asiklik dari guanosin yang dapat menghambat replikasi VZV dan merupakan prodrug yang memerlukan tahapan fosforilasi untuk menjadi bentuk aktifnya. Thymidine kinase (TK) akan mengonversi acyclovir menjadi acyclovir monophosphate. Kemudian enzim seluler akan mengatalisis tahapan difosforilasi dan trifosforilasi sehingga menghasilkan bentuk acyclovir triphosphate. Acyclovir triphosphate dapat menghambat sintesis DNA virus dan bersaing dengan deoxyguanosine triphosphate sebagai substrat untuk DNA polimerase virus.[4] Penggunaan acyclovir sebagai antivirus pada infeksi VZV saat ini memiliki tantangan tersendiri terutama pada individu imunodefisiensi. Sekitar 30% kasus infeksi VZV dilaporkan mengalami resistansi acyclovir karena adanya mutasi yang mengubah aktivitas enzimatik virus.[7]
Pilihan antivirus lain telah tersedia untuk mengatasi infeksi VZV yang resistan terhadap acyclovir seperti foscarnet, penciclovir, cidofovir, dan brincidofovir. Namun, beberapa efek samping dan toksisitas dilaporkan pada penggunaan terapi lini kedua dan ketiga tersebut.[8] Saat ini, terapi agen antivirus golongan analog nukleosida dan non nukleosida yang baru sudah dikembangkan sebagai alternatif pada infeksi VZV yang tidak dapat menerima terapi agen antivirus yang telah tersedia.[9]
Artikel ini bertujuan untuk mengetahui mekanisme resistansi VZV terhadap acyclovir dan dapat menganalisa kemungkinan resistansi pada kasus infeksi VZV sehingga dapat diberikan terapi yang tepat. Pemberian terapi yang tepat diharapkan dapat mencegah komplikasi infeksi virus lebih lanjut.
Virus Varisela Zoster
Varicella zoster virus adalah virus alpha-herpes pada manusia yang menyebabkan cacar air (varisela) sebagai infeksi primer dan herpes zoster (shingles) sebagai reaktivasi dari infeksi varisela primer.[10,11] Varisela primer umumnya merupakan penyakit self-limited pada anak-anak yang imunokompeten.[11] Virus varisela merupakan virus deoxyribonucleic acid (DNA) untai ganda dan terdiri dari 68 open reading frame (ORF) unik, tersusun dari kurang lebih 125.000 bp genom, berselubung dan berdiameter 80-120 nm.[10] Virus ini meng-kode 70-80 protein, salah satunya enzim thymidine kinase (TK) yang dapat memfosforilasi asiklovir menjadi bentuk aktifnya sehingga dapat menghambat replikasi DNA virus (Gambar 1).[6]
Virus neurotropik ini menjadi laten setelah infeksi primer terutama di neuron ganglion otonom perifer pada seluruh neuroaksis termasuk akar dorsal ganglion, ganglion saraf kranial seperti ganglion trigeminal dan ganglion otonom termasuk yang ada di sistem saraf enterik. Virus varisela zoster laten dapat teraktifasi kembali sehingga menyebabkan herpes zoster. Reaktivasi virus ini dapat terjadi pada pasien yang berusia lebih dari 50 tahun, memiliki kondisi gangguan kekebalan tubuh, konsumsi obat-obatan imunosupresif, HIV/AIDS, keganasan, stres psikologis, menjalani transplantasi sumsum tulang atau organ, atau pada kondisi lain yang menyebabkan penurunan kekebalan tubuh.[6,10,11]
Masa inkubasi VZV berkisar 10-21 hari setelah terpapar, dengan rerata masa inkubasi sekitar 14-16 hari. Siklus hidup VZV dimulai dengan masuknya virus ke dalam sel inang melalui terjadinya fusi dari partikel virus dengan membran plasma atau endositosis. Glikoprotein yang terdapat pada permukaan sel virus memiliki peranan penting terjadinya interaksi virus dengan sel inang termasuk penyebaran virus dari sel ke sel. Glikoprotein E akan berinteraksi dengan insulin degrading enzyme (IDE) untuk memediasi masuknya virus ke dalam sel.[7,12,13,14] Sedangkan kombinasi gH dan gL atau gB dan gE diperlukan untuk penggabungan antar sel. Glikoprotein H dan M terutama diperlukan untuk penyebaran dari sel ke sel, sedangkan gK akan memfasilitasi pembentukan sinsitia.[14] Virion akan dilepaskan ke sitoplasma setelah berpenetrasi ke membran sel dan kapsid yang berikatan dengan protein tegumen akan ditransportasikan ke nukleus. Asam nukleat virus terpisah dari selubung proteinnya pada nukleoplasma. Asam nukleat tersebut kemudian masuk ke dalam nukleus dan menginisiasi proses transkripsi dan replikasi virus.[7]

Gambar 1. (A-B) Morfologi struktur VZV. Virion terdiri dari glikoprotein, selubung lemak, tegumen dan kapsid nukleus yang melindungi bagian inti yaitu genom DNA untai ganda.[6]
Epidemiologi
Varisela atau cacar air adalah penyakit yang disebabkan karena infeksi virus varisela zoster yang dapat ditularkan melalui udara atau kontak langsung dengan cairan vesikel dari penderita varisela. Penyakit ini umumnya jinak dan dapat sembuh sendiri serta lebih sering terjadi pada anak- anak dibandingkan dewasa.[15,16] Populasi berusia di atas 13 tahun memiliki peningkatan risiko 2,2 kali lipat tertular varisela derajat sedang sampai berat dibandingkan dengan usia yang lebih muda. Terdapat 15 kasus tiap 1000 orang terjadi per tahun dengan lebih dari setengahnya berusia di bawah 5 tahun dan sekitar 85% kasus terjadi sebelum pubertas.[13,17] Meskipun infeksi virus ini relatif jinak namun tetap berpotensi menyebabkan kematian. Dilaporkan di wilayah Inggris dan Wales, tingkat kematian pada pasien usia 15-44 tahun adalah 20 kali lebih tinggi dari kelompok usia 5–14 tahun. Terdapat 0,94% kematian per 100.000 penduduk terdapat pada kelompok usia muda 5–14 tahun dan 20,06% pada mereka yang berusia lebih tua 15-44 tahun.[15,16] Pada pasien wanita hamil yang terinfeksi VZV dapat mengalami komplikasi dan konsekuensi yang cukup berat bagi janin.[15] Secara umum varisela dapat terjadi di seluruh dunia tetapi lebih banyak didapati pada negara dengan iklim sedang dibanding dengan tropis. Laporan kasus varisela pada negara dengan iklim sedang umumnya terjadi saat musim dingin dan semi.[13,17,18] Hal ini dikaitkan dengan aktivasi virus yang mulai terjadi pada temperatur 5°C hingga 20°C.[19]
Insidensi herpes zoster terjadi pada lebih dari 1 juta orang di Amerika Serikat dan memiliki insidensi berkisar 20% selama hidup.[11] Secara global dilaporkan, rerata insiden herpes zoster berkisar 2-4 kasus per 1000 orang per tahunnya. Insidensi dan keparahan herpes zoster dapat meningkat dan sering mengalami komplikasi berupa infeksi diseminata dan keterlibatan sistemik yakni pneumonia, hepatitis atau ensefalitis pada pasien dengan imunitas yang terganggu. Insidensi herpes zoster mencapai setidaknya 10% pada individu yang mengalami malignansi khususnya limfoma. Terdapat 30-40% pasien yang terinfeksi VZV selama tahun pertama setelah transplantasi pada pasien yang menerima terapi sitotoksik atau imunosupresan khususnya pasien transplantasi sumsum tulang belakang. Risiko terjangkit infeksi VZV meningkat 10 kali lebih tinggi dibandingkan dengan populasi normal dan dapat menjadi infeksi diseminata yang kronis pada pasien yang terinfeksi HIV.[13]
Penyakit yang Disebabkan oleh Virus Varisela Zoster
a. Varisela
Varisela atau dikenal dengan chickenpox merupakan penyakit infeksi yang disebabkan VZV. Gejala infeksi virus ini umumnya muncul dua minggu setelah pajanan seperti demam yang tidak tinggi, menggigil, gatal, sakit kepala, malaise, nyeri perut, mual, muntah, batuk, koriza dan radang tenggorokan selama 2-3 hari. Fase prodromal ini akan berkurang sekitar 7-14 hari setelah pajanan VZV.[11,12,13] Selanjutnya, diikuti dengan munculnya ruam berupa makula eritematosa pada wajah dan kulit kepala yang kemudian menyebar ke badan dan bagian distal dari ekstremitas atas dan bawah (Gambar 2). Dua belas jam kemudian makula akan berkembang menjadi papul berukuran 1-3 mm, vesikel dan kemudian pustul. Lesi baru lainnya dapat muncul sekitar 3-5 hari. Lesi vesikel diatas lesi eritematosa dideskripsikan sebagai gambaran tetesan embun di atas kelopak mawar. Pustul membentuk krusta setelah 6-7 hari dan lesi akan sembuh tanpa disertai skar.[12] Krusta terjadi dalam beberapa hari dan akan terjadi penyembuhan sempurna setelah 1 bulan. Penyembuhan lesi biasanya terjadi dalam beberapa fase yang berbeda dalam satu regio.[11] Derajat nyeri dan luas lesi berbeda pada setiap individu. Pembengkakan, eritema luas, demam lebih dari 4 hari, erupsi lesi baru yang memanjang atau keterlambatan penyembuhan menjadi penanda adanya infeksi sekunder bakteri atau kerusakan imunitas seluler.[12] Gejala varisela dapat muncul dalam berbagai spektrum mulai dari gejala ringan dengan sedikit ruam hingga berat dengan jumlah vesikel lebih dari 1000 vesikel. Penyakit varisela didapati 25 kali lebih berat dan fatal bila dialami orang dewasa dibandingkan dengan anak-anak. Orang dewasa yang menderita varisela rentan berkembang menjadi pneumonia varisela namun komplikasi ini umumnya memiliki respon yang cepat terhadap pengobatan antivirus.[13]
Penularan varisela yang paling tinggi terjadi pada fase mulai terbentuk vesikel. Penularan akan menurun setelah memasuki tahap pustular dan sudah tidak menular apabila semua lesi telah menjadi krusta.[13,20] Antibodi imunoglobulin G (IgG), IgM dan IgA pada varisela muncul 2-5 hari setelah onset ruam dan mencapai puncak pada minggu kedua dan ketiga. Kemudian titer IgG akan menurun perlahan dan bertahan pada level yang rendah. Level antibodi IgG akan meningkat secara cepat dan menjadi lebih tinggi dibandingkan pada infeksi primer apabila terjadi herpes zoster setelahnya.[13]

Gambar 2. (A-B). Gambaran klinis varisela pada anak berusia 4 tahun. Tampak lesi kulit dalam berbagai tahapan disertai erosi dan ekskoriasi. (C). Gambaran vesikel lesi awal berupa gambaran tetesan embun di atas kelopak mawar. (D). Lesi yang sudah menjadi krusta.[4]
Varisela dapat menimbulkan komplikasi pada sistem saraf pusat yakni cerebellar ataxia yang dapat sembuh sendiri serta dapat terjadi komplikasi lebih serius ensefalitis. Pada pasien imunodefisiensi yang terinfeksi varisela dapat mengalami komplikasi yang fatal seperti perjalanan penyakit yang memanjang, demam tinggi, ruam yang ekstensif yang kemudian berkembang menjadi hemoragik, pneumonia, hepatitis dan ensefalitis. Varisela maternal pada minggu ke-8 hingga 26 kehamilan berpotensi menyebabkan gangguan pada janin yang didapati pada sekitar 2% kasus. Gangguan tersebut meliputi lesi kulit, kerusakan sistem saraf pusat dan otonom, gangguan okular dan kecacatan. Mortalitas pada neonatus dengan ibu yang menderita varisela dilaporkan sebesar 30%. Bayi yang lahir dengan sindrom varisela kongenital biasanya tidak dapat bertahan dan apabila bertahan akan muncul herpes zoster pada awal kehidupan.[20]
b. Herpes Zoster
Herpes zoster merupakan penyakit sekunder VZV yang disebabkan reaktivasi VZV yang didapatkan saat menderita varisela dan mengalami latensi setelahnya.[13,20] Reaktivasi VZV dari latensi dapat terjadi karena adanya antibodi VZV pada sirkulasi dan titer VZV yang tinggi. Lesi herpes zoster dapat berupa vesikel yang nyeri dan gatal tersebar sesuai dermatom dan umumnya bersifat unilateral.[10,11,20] Spektrum penyakit ini bervariasi dari nyeri tanpa disertai ruam sampai dengan ruam ringan dan ruam berat disertai diseminata. Individu yang mengalami herpes zoster dapat menularkan VZV kepada individu lainnya dan menyebabkan individu tersebut menderita varisela sebagai penyakit primer yang diinduksi VZV. Herpes zoster dapat muncul tanpa disertai ruam yang disebut zoster sine herpete dengan manifestasi berupa sindrom nyeri terbatas pada dermatom yang bersifat unilateral, ensefalitis, manifestasi neurologis atau gangguan gastrointestinal.[20,21]

Gambar 3. Gambaran klinis herpes zoster berupa vesikel berkelompok pada satu area dermatom.16
Lebih dari 90% pasien herpes zoster mengalami gejala prodromal berupa nyeri hebat pada satu ganglion sensoris atau dermatom yang berkembang menjadi ruam zoster. Lesi khasnya berupa ruam dermatomal yang muncul sebagai vesikel eritematosa diikuti krusta (Gambar 3). Pasien akan mengeluh gejala konstitusional yakni rasa terbakar, nyeri atau gatal pada area sebelum munculnya erupsi vesikel. Herpes zoster biasanya hanya melibatkan satu dermatom dan tidak melewati garis tengah tubuh. Lokasi predileksi yang paling sering yakni regio torakalis. Lesi kulit umumnya muncul pada area dermatom yang paling parah ketika terjadi infeksi VZV primer namun dapat juga menyebar ke dermatom sekitarnya.[11,12] Diagnosis herpes zoster dapat ditegakkan melalui temuan klinis, jika diperlukan pemeriksaan laboratorium seperti pengecatan Tzank dapat digunakan untuk mengonfirmasi diagnosis penyakit ini.[12]
Pasien yang menderita herpes zoster dapat mengalami gangguan hingga kehilangan penglihatan karena adanya nekrosis retina progresif yang dapat berkembang menjadi kebutaan terutama pada pasien dengan imunodefisiensi seperti infeksi HIV.[20] Tidak hanya itu saja herpes zoster pada pasien dengan gangguan sistem kekebalan tubuh dapat menyebabkan oftalmitis disertai keratitis. Komplikasi okular dari zoster oftalmika juga dapat terjadi pada jika virus juga bereplikasi hingga ke-nervus oftalmikus. Vaskulopati akibat VZV yang disertai dengan herpes zoster dapat muncul sebagai arteritis pada arteri serebri. Kondisi serius ini menimbulkan beberapa gejala seperti kerusakan serebrovaskular, gangguan penglihatan hingga kebutaan dan transient ischemic attack (TIA).[12,20,21] Herpes zoster juga dapat menyebabkan kerusakan serabut saraf pada bagian tengah dan bawah dermis yang dapat dideteksi dengan teknik impregnasi perak. Denervasi parsial dapat bertahan selama lebih dari satu tahun yang khas terjadi pada pasien post herpetic neuralgia sebagai sindrom nyeri neuropatik yang sering terjadi setelah herpes zoster. Post herpetic neuralgia umumnya terjadi apabila terdapat nyeri dermatomal yang lebih lama pada erupsi sebelumnya, terdapat nyeri akut herpes zoster yang berat dan ruam zoster yang lebih lama dari biasanya.[13,20] Komplikasi herpes zoster lainnya yang dapat muncul yakni timbulnya lesi ulseratif (Gambar 4), adanya vesikel pada beberapa dermatom atau erupsi bilateral, vesikel yang dalam dan merusak kulit sehingga menyebabkan timbulnya skar (Gambar 5), kelemahan otot, palsy pada saraf fasialis yang reversibel serta infeksi organ dalam meliputi saluran gastrointestinal, paru-paru dan otak (ensefalitis).[1]

Gambar 4. (A-B) Gambaran lesi herpes zoster ulseratif tampak nekrosis, erosi dan ulserasi pada herpes zoster.[1]
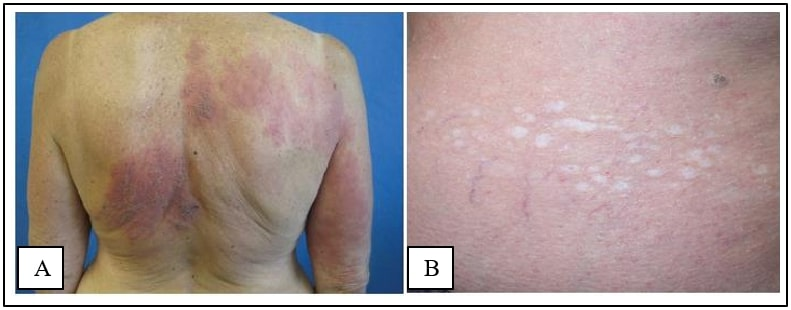
Gambar 5. (A). Erupsi bilateral pada herpes zoster. (B). Skar setelah infeksi herpes zoster.[1]
Mekanisme Kerja Acyclovir
Acyclovir merupakan analog nukleosida yang menjadi lini pertama terapi penyakit varisela.[22] Acyclovir bekerja menghambat replikasi DNA dari VZV dan virus herpes simpleks pada manusia melalui perannya sebagai terminator rantai DNA. Acyclovir akan mengalami fosforilasi oleh enzim TK dari virus untuk menjadi bentuk aktif yaitu acyclovir triphospat yang merupakan inhibitor kompetitif dari polimerase DNA virus dan menyebabkan terminasi rantai DNA virus.[8,23,24] Terapi acyclovir dapat digunakan untuk jangka pendek pada infeksi herpes primer dan rekuren, maupun untuk terapi jangka panjang pada herpes genital rekuren, terapi herpes zoster dan varisela.[22]
Formulasi acyclovir dalam bentuk injeksi intravena diindikasikan untuk pengobatan herpes simplex virus (HSV) berat seperti ensefalitis, herpes neonatal dan infeksi VZV.[22] Acyclovir intravena memiliki penetrasi yang tinggi pada jaringan dan cairan termasuk cairan serebrospinal karena dapat berikatan kuat dengan plasma protein. Sedangkan, acyclovir oral hanya memiliki bioavaibilitas berkisar 15- 30% dari acyclovir intravena sehingga memerlukan dosis pemberian yang lebih sering untuk memberikan efek terapi pada plasma darah. Waktu paruh plasma acyclovir berkisar 2-3 jam pada pasien dewasa dengan fungsi ginjal yang normal. Acyclovir dapat ditoleransi dengan baik pada tubuh manusia, tidak menimbulkan toksisitas dan efek sampingnya minimal. Acyclovir disekresikan melalui ginjal oleh filtrasi glomerulus dan sekresi tubulus sehingga dapat menyebabkan nefrotoksisitas terutama jika diberikan melalui infus intravena yang cepat pada pasien yang mengalami dehidrasi atau memiliki gangguan ginjal. Konsentrasi tinggi acyclovir pada serum dapat menyebabkan delirium dan gejala neurologis reversibel lainnya pada orang tua atau pasien dengan gangguan fungsi ginjal. Penggunaan acyclovir pada masa kehamilan tergolong dalam obat kategori B, yang berarti penelitian pada hewan, obat tidak menunjukkan efek teratogenik namun tidak ada bukti yang adekuat dan studi yang terkontrol penggunaan obat ini pada ibu hamil. Beberapa studi menyatakan bahwa penggunaan acyclovir tidak berhubungan dengan peningkatan prevalensi kecacatan lahir.[23]
Mekanisme Resistansi Acyclovir
Pengobatan acyclovir untuk infeksi VZV tidak menghasilkan resistansi acyclovir pada orang yang imunokompeten.[9] Resistansi acyclovir pada infeksi VZV dapat dijumpai pada pasien imunodefisiensi yang mengalami infeksi VZV lebih berat sehingga diperlukan terapi dengan acyclovir dalam jangka panjang.[8,22] Komplikasi terkait infeksi VZV dan resistansi terhadap antivirus merupakan faktor yang berperan pada infeksi persisten pasien hemato-onkologi.[9] 27% pasien hemato-onkologi yang mendapatkan hematopoietic stem cell transplantation (HSCT) menderita infeksi VZV persisten akibat mutase virus yang berhubungan dengan resistansi acyclovir.[22] Satu kasus melaporkan, pasien HIV dengan infeksi VZV kutaneus diseminata yang mendapatkan acyclovir memberikan respons perbaikan namun mengalami rekurensi setelah pasien menerima acyclovir dengan dosis 0,4-4 gram selama 1-5 bulan.[23]
Infeksi VZV yang resistan terhadap acyclovir harus dicurigai pada lesi khas zoster yang menetap setelah 10 hari penggunaan acyclovir sesuai dosis terapi.[8] Resistansi acyclovir pada isolat VZV sebagian besar diakibatkan oleh mutasi pada TK virus (gen ORF36) dan sebagian kecil disebabkan oleh mutasi polimerase DNA virus (gen ORF28). Genome VZV memiliki kandungan guanin-kitosin yang lebih rendah (46%) daripada pada HSV (68%) dan hanya terdapat sedikit homopolimer pada gen ORF36. Isolat VZV yang resistan terhadap acyclovir mengalami delesi nukleotida pada homopolimer sehingga menyebabkan urutan nukleotida yang membentuk asam amino spesifik (kodon) terhenti pada enzim TK. Substitusi nukleotida nonsinonim juga menyebabkan resistansi acyclovir menyebar pada gen ORF36 namun lebih sering terjadi pada ATP-binding dan nucleoside-binding. Substitusi asam amino polimerase DNA banyak ditemukan pada proses katalisis dan mekanisme kerja enzim yang menyebabkan resistansi pada acyclovir.[22,25]
Resistansi VZV terhadap acyclovir dapat terjadi melalui beberapa mekanisme yakni defisiensi enzim TK yang menjadi mekanisme yang sering didapati, produksi TK yang rendah (penurunan aktivitas enzimatis hanya 1-5% dari aktivitas normal), perubahan TK dan perubahan fenotip polimerase DNA (perubahan aktivitas enzim).[26,27,28] Mutasi pada TK akan menghilangkan atau menurunkan aktivitas enzim sehingga tidak dapat melakukan fosforilasi timidin untuk replikasi virus. Defisiensi dan produksi TK yang rendah dapat menyebabkan reaktivasi virus wild strain. Individu imunodefisiensi dengan infeksi strain virus yang resistan terhadap acyclovir memiliki risiko lebih tinggi mengalami reaktivasi virus pada ganglia saraf.[26,29,30]
Deteksi Resistansi Acyclovir
Beberapa komplikasi dapat terjadi pada berbagai organ setelah reaktivasi VZV antara lain kulit, organ dalam, neurologis dan okular. Pengobatan standar pada infeksi VZV yang sering diberikan adalah acyclovir analog guanosin namun resistansi terhadap obat ini semakin banyak dilaporkan. Penegakan resistansi terhadap acyclovir secara lebih dini diperlukan agar terapi alternatif lain dapat tepat waktu diberikan.[31] Infeksi VZV yang dicurigai resistan terhadap antivirus dapat dikonfirmasi dengan pemeriksaan isolat klinis melawan agen antivirus pada kultur sel (pemeriksaan fenotip) dan dengan identifikasi mutasi spesifik yang menyebabkan resistansi obat secara langsung pada sampel klinis (pemeriksaan genotip).[8,26]
a. Pemeriksaan Fenotip
Kerentanan VZV terhadap obat antivirus dapat dideteksi dengan pemeriksaan plaque reduction assay (PRA) menggunakan sel fibroblastik. Virus yang diduga mengalami resistansi dikembangkan melalui kultur pada sel tersebut.[22]
Plaque reduction assay (PRA) merupakan metode fenotipik yang menjadi standar untuk menentukan kerentanan isolat virus terhadap antivirus. Sel dipaparkan secara konstan dengan inokulum virus pada pemeriksaan ini. Virus kemudian dibiarkan tumbuh pada dilusi serial obat selama 2-3 hari sebelum dilakukan fiksasi dan pewarnaan sel. Efek sitopatik atau plak virus kemudian dihitung menggunakan mikroskop. Konsentrasi obat yang menurunkan 50% jumlah plak dibandingkan dengan kontrol (yang tidak ditambahkan dengan agen antivirus) didefinisikan sebagai konsentrasi efektif 50% (EC50). Jika nilai EC50 ≥9 µM, menunjukkan resistansi terhadap acyclovir. Peningkatan nilai EC50 ≥4 kali pada strain yang dicurigai dapat diartikan juga sebagai resistansi VZV terhadap acyclovir. Pemeriksaan PRA memiliki beberapa kelemahan seperti sulitnya mendapatkan spesimen klinis yang cukup pada kultur sel, waktu pertumbuhan isolat pada kultur yang lama, subjektivitas dalam penghitungan plak dan populasi virus heterogen yang tumbuh pada kultur memungkinkan terjadinya bias.[8,22,31]
Objektivitas pemeriksaan PRA dapat ditingkatkan dengan kombinasi metode lainnya seperti deteksi antigen spesifik (ezyme-linked immunosorbent assay (ELISA), flow cytometry atau pewarnaan imunoperoksida dan dengan deteksi DNA (hibridisasi dan real time PCR).[8,22,32]
b. Pemeriksaan Genotip
Pemeriksaan genotip berdasarkan amplifikasi gen VZV yang resistan terhadap obat- obatan tertentu menggunakan PCR merupakan pemeriksaan yang lebih cepat dan sensitif dibandingkan dengan pemeriksaan fenotip.[8] Pemeriksaan ini menggunakan identifikasi mutasi spesifik dengan pengurutan DNA. Pengurutan dideoksi standar dapat mendeteksi mutasi resistan yang muncul ketika terjadi pada 20% dari total populasi sehingga diperkirakan viral load paling sedikit 1000 kopi/ml sampel klinis untuk mendapatkan profil genotipik yang valid. Mutasi menyebabkan resistansi terhadap analog nukleosida terjadi pada gen VZV ORF 36 yang merupakan pengkodean dari TK dan pada gen VZV ORF 28 yang merupakan pengkodean dari polimerase DNA. Interprestasi hasil pemeriksaan genotip dibandingkan dengan mutase yang telah tercatat pada polimorfisme natural atau resistansi obat yang telah terkonfirmasi pada literatur.[22,33]
Enzim TK VZV di-enkode oleh ORF36 dan sangat penting pada tahap awal dalam fosforilasi acyclovir menjadi bentuk trifosfat aktifnya. Analisis pengurutan TK VZV oleh sebab itu dapat digunakan dalam sebagai metode diagnostik klinis rutin. Analisis pengurutan TK VZV membutuhkan waktu yang jauh lebih pendek dibandingkan dengan pemeriksaan fenotip.
Analisis ini dapat diterapkan pada sampel yang mengandung segala bentuk virus DNA dan pembacaannya tidak terhambat oleh variabilitas antar peneliti.[31]
Alternatif Terapi Antivirus pada Infeksi VZY yang Resistan terhadap Acyclovir
a. Penciclovir dan Famciclovir
Penciclovir adalah turunan guanin asiklik yang menyerupai acyclovir dalam struktur kimia, mekanisme kerja dan spektrum aktivitas antivirus. Penciclovir akan mengalami fosforilasi oleh TK virus seperti pada acyclovir kemudian menjadi bentuk trifosfat oleh enzim seluler. Penciclovir trifosfat dapat menghambat sintesis DNA virus melalui penghambatan kompetitif dengan polimerase DNA virus. Penciclovir trifosfat memiliki karakter yang berbeda dengan acyclovir trifosfat yakni bukan merupakan terminator rantai obligat dan dapat bergabung ke dalam rantai DNA yang memanjang. Konsentrasi intraseluler penciclovir trifosfat lebih tinggi dibanding dengan acyclovir trifosfat. Waktu paruh penciclovir trifosfat adalah 7 jam dan acyclovir hanya 1 jam pada sel yang terinfeksi VZV namun penciclovir trifosfat memiliki afinitas pada polimerase DNA virus yang lebih rendah. Penciclovir tidak dimetabolisme namun dieliminasi di urine dengan waktu paruh eliminasi sekitar 2 jam setelah pemberian intravena. Penciclovir intravena tidak diperdagangkan secara komersial. Famciclovir dikembangkan sebagai formulasi oral karena absorbsi pensiklovir yang rendah. Famciclovir merupakan prodrug dari penciclovir yang dapat diberikan dengan dosis 500 mg setiap 8 jam pada infeksi VZV. Pemberian famciclovir harus dipertimbangkan kembali pada pasien dengan klirens kreatinin <60ml/min. Efek samping yang paling sering muncul adalah sakit kepala dan mual.[23,34]
Pemeriksaan in vitro pada mutasi polimerase DNA menunjukkan bahwa acyclovir dan penciclovir memiliki genotip VZV resistan obat yang berbeda. Mutasi TK lebih banyak terjadi pada resistansi acyclovir sedangkan mutasi polimerase DNA lebih banyak terjadi terjadi pada resistansi penciclovir. Penciclovir dapat tetap aktif melawan herpes virus yang mengalami mutasi polimerase DNA dan TK virus yang resistan terhadap acyclovir. Temuan ini mengindikasikan bahwa interaksi antara TK VZV dengan penciclovir atau acyclovir dan antara polimerase virus dengan penciclovir trifosfat atau acyclovir berbeda. Hal ini menggambarkan perbedaan strain VZV yang resistan terhadap acyclovir dan penciclovir. Penciclovir oleh sebab itu dapat menjadi salah satu terapi pilihan yang dapat digunakan pada infeksi VZV yang resistan terhadap acyclovir.[8]
b. Foscarnet
Foscarnet merupakan analog pirofosfat, oksianion fosfor yang ditemukan dalam molekul DNA. Analog ini bertindak seperti molekul pirofosfat yaitu berikatan secara selektif dan reversibel pada tempat ikatan enzim polimerase DNA virus sehingga dapat menghambat rantai DNA untuk tidak memanjang.[38]
Foscarnet tersedia dalam bentuk sediaan injeksi intravena yang dosis dan kecepatan pemberian ditentukan berdasarkan usia dan berat pasien serta infeksi virus tertentu.[23,25] Dosis foscarnet dapat diberikan sebesar 40 mg/kg secara intravena setiap 8 jam sampai lesi sembuh.[4] Penelitian terhadap isolat resistan acyclovir yang diambil dari 10 pasien AIDS menunjukkan hasil yang sensitif pada penggunaan foscarnet. Penelitian menunjukkan pemberian foscarnet intravena memberikan keberhasilan terapi pada 10 dari 13 pasien AIDS dengan infeksi VZV yang resistan terhadap acyclovir.[23]
Efek samping dari foscarnet yang paling menonjol adalah mual, gangguan keseimbangan elektrolit dan penurunan fungsi ginjal. Efek samping mual yang terjadi selama pemberian infus foscarnet dapat diatasi dengan pemberian antiemetik dan memperlambat kecepatan infus obat. Selain mual, insufisiensi ginjal adalah kejadian yang relatif paling sering dilaporkan dari penggunaan foscarnet. Gangguan fungsi ginjal akut yang terjadi bersifat reversibel dan sangat penting untuk diatasi apabila teridentifikasi. Foscarnet memengaruhi kerja sel tubulus ginjal melalui mekanisme sitotoksik langsung dan tingkat toksisitasnya berkorelasi dengan dosis yang diberikan. Foscarnet juga dapat menyebabkan nefropati kristal dengan pengendapan kristal di kapiler glomerulus. Kristal ini sering kali merupakan campuran garam natrium dan kalsium. Oleh karena itu, perlu melakukan pemantauan berkala pada fungsi ginjal untuk pasien yang menjalani terapi dengan foscarnet. Cedera tubulus ginjal ditandai dengan adanya peningkatan kreatinin plasma dalam 1 sampai 2 minggu pemberian foscarnet. Foscarnet pada beberapa kasus juga dapat menyebabkan diabetes insipidus nefrogenik.[23,37]
Gangguan keseimbangan elektrolit seperti hipokalsemia dan hipomagnesemia juga sering didapati pada pasien pengguna foscarnet. Hipokalsemia mungkin disebabkan oleh pembentukan komplek dari foscarnet dengan ion kalsium ataupun karena kondisi hipomagnesemia yang diinduksi foscarnet sehingga mengakibatkan hipokalsemia tersebut maupun hipokalemia. Pemantauan keseimbangan elektrolit serta melihat hasil EKG awal sebelum pengobatan pada pasien pengguna foscarnet perlu dilakukan untuk mencegah terjadinya efek samping sistemik seperti aritmia jantung.[37]
Penggantian kalium dan magnesium harus diberikan untuk mencegah efek samping tersebut.[8,37]
c. Cidofovir
Cidofovir merupakan analog nukleotida monofosfat yang sudah memiliki satu gugus fosfat tunggal sehingga tidak memerlukan kerja kinase virus untuk fosforilasi awal. Kinase seluler memfosforilasi molekul menjadi cidofovir difosfat yang dapat menghambat polimerase DNA virus.[35] Polimerase DNA virus menunjukkan afinitas 25-50 kali lebih besar terhadap cidofovir difosfat dibandingkan dengan polimerase seluler host sehingga menghambat replikasi DNA virus. Cidofovir memiliki aktivitas in vitro melawan semua jenis herpes virus pada manusia.[36,38] Cidofovir dapat diberikan secara intravena dengan dosis 5 mg/kg sekali seminggu selama 3-4 minggu.[26,38] Konsentrasinya pada sel ginjal 100 kali lebih tinggi dibandingkan di jaringan lainnya sehingga dapat menyebabkan kerusakan tubulus proksimal.[8,36,38]
Toksisitas pada ginjal ditandai dengan adanya proteinuria dan glikosuria. Prehidrasi intravena dan pemberian probenesid oral dapat dilakukan untuk mengurangi nefrotoksisitas akibat cidofovir. Terapi cidofovir juga dikaitkan dengan supresi sumsum tulang dan uveitis.[8,35,38]
d. Brincidofovir
Brincidofovir yang tersedia dalam bentuk sediaan oral merupakan konjugat lemak cidovofir analog nukleotida. Brincidofovir secara in vitro memiliki aktivitas antivirus melawan DNA virus untai ganda termasuk VZV, virus herpes, virus polioma, adenomavirus, human papilloma viruses dan poxvirus. Enzim TK virus tidak diperlukan untuk aktivasi senyawa ini. Keberhasilan penggunaan terapi brincidofovir telah dilaporkan pada manajemen terapi infeksi VZV yang resistan terhadap cidofovir dan asiklovir pada pasien intoleransi foscarnet.[8]
Penelitian menunjukkan brincidofovir memiliki waktu paruh intraseluler yang cukup panjang pada penggunaan dosis oral dua kali seminggu. Meskipun demikian obat ini bukanlah substrat untuk transporter anion organik manusia sehingga dapat menyebabkan toksisitas pada ginjal. Ikatan fosfat ester lipid dari brincidofovir dipisahkan oleh fosfolipase intraseluler yang melepaskan cidofovir serta kemudian dikonversikan menjadi cidovofir difosfat oleh kinase seluler. Brincidofovir menghasilkan konsentrasi cidovofir difosfat 100 kali lebih tinggi dibandingkan dengan cidofovir. Cidofovir difosfat juga 300-400 kali lebih aktif melawan herpes simpleks virus-1 (HSV-1) dan cytomegalovirus (CMV) dibandingkan cidofovir. Konsentrasi efektif cidofovir disfosfat (EC50) terhadap VZV adalah 1000 kali lebih rendah dari nilai EC50 cidofovir sehingga penggunaan brincidofovir daapt secara efektif mengatasi resistansi cidovofir.[8]
Peningkatan enzim hati dilaporkan pada individu yang diterapi dengan brincidofovir. Perubahan histopatologis tidak didapatkan pada peningkatan enzim ini pada studi preklinik. Gangguan hepatobilier juga dapat terjadi pada terapi menggunakan obat ini. Temuan yang paling sering yakni hiperbilirubinemia. Efek samping gastrointestinal berupa diare, nyeri abdomen dan muntah dapat muncul pada pemberian brincidofovir lebih dari 200 mg dua kali perminggu selama lebih dari 2-4 minggu.[8]
Terapi yang Sedang Dikembangkan pada Infeksi Varisela Zoster Virus
Resistansi VZV terhadap asiklovir telah dilaporkan pada beberapa kasus di seluruh dunia terutama pada individu imunodefisiensi. Pilihan antivirus yang telah berkembang saat ini seperti foscarnet, penciclovir, cidofovir dan brincidofovir dapat menjadi pilihan terapi pada kasus tersebut namun efek samping atau toksisitas yang muncul akibat terapi tersebut menjadi keterbatasan dalam penggunaannya sebagai terapi pada infeksi VZV. Beberapa kandidat agen antivirus dikembangkan untuk mengatasi hal tersebut.
a. Novel Nucleoside Analog (NA)
Terdapat 3 NA yang saat ini sedang aktif dikembangkan untuk melawan VZV yakni valnivudine hydrochloride (FV-100), valomaciclovir stearate (EPB-348) dan N- methanocarbathymidine (N-MCT, NN-001).
1. Valnivudine hydrochloride (FV-100)Valnivudine hydrochloride saat ini termasuk antiviral yang masih dikembangkan. Obat ini merupakan valine ester dari bicyclic nucleoside analogues (BCNA) Cf1743. Bicyclic nucleoside analogues mampu menghambat aktivitas anti VZV melalui cara yang sama sekali berbeda dengan analog nukleosida lainnya. Studi preklinik menunjukkan nilai EC50 BCNA adalah 300 kali lebih poten dalam menghambat VZV dibandingkan dengan acyclovir. Berbeda dengan acyclovir yang bekerja dengan melakukan terminasi rantai DNA ketika difosforilasi menjadi bentuk trifosfat, sedangkan BCNA masih belum diketahui apakah akan diubah menjadi bentuk trifosfat, serta bentuk mana yang merupakan metabolit aktif maupun bagaimana mekanisme kerjanya secara pasti.[35,39,40] Studi keamanan valnivudine menunjukkan obat ini akan dengan cepat mengalami konversi menjadi metabolit CF-1743 yang secara rendah akan dieksresikan melalui ginjal. Profil farmakokinetika valnivudine pada pasien lanjut usia maupun pasien anak-anak adalah serupa. Studi fase I valnivudide menunjukkan bahwa pemberian secara oral dengan dosis 1 kali per hari dapat ditoleransi dengan baik dan terbukti dapat mempertahankan konsentrasi in vivo obat di atas EC50.[35,40] Sedangkan studi keamanan dan efikasi valnivudine dibandingkan valasiklovir untuk mereduksi nyeri post herpetic menunjukkan bahwa penggunaan valnivudine 200 mg atau 400 mg sehari sekali merupakan pilihan terapi yang potensial untuk mengatasi nyeri post herpetic dibandingkan penggunaan valaciclovir 1000 mg 3 kali sehari.[35]
2. ValomaciclovirValomaciclovir adalah prodrug diester yang dengan cepat akan mengalami konversi secara in vivo menjadi acyclic guanosine analogue analog (R)-9-[hydroxy-2-(hydroxymethyl)butyl]guanine H2G. Aktivasi H2G terjadi mirip dengan asiklovir melalui trifosforilasi oleh TK yang menghasilkan polimerase DNA virus. Obat ini paling efektif melawan HSV-1, HSV-2, VZV dan EBV. Aktivitas in vitro H2G melawan VZV lebih tinggi dibanding acyclovir.40 Valomaciclovir merupakan prodrug omaciclovir dan bentuk trifosfat omaciclovir aktif memiliki waktu paruh intraseluler lebih lama (4-14 jam) dibanding trifosfat acyclovir (1-2 jam), serta memiliki bioavailabilitas yang baik sehingga dosis yang diberikan dapat lebih minimal.[39]
Studi efikasi terapi herpes zoster dengan menggunakan valomaciclovir dan valaciclovir untuk pasien immunocompetent menunjukkan valomaciclovir yang diberikan dengan dosis 2000 mg dan 3000 mg 1 kali sehari efektif untuk mengurangi lesi herpes di hari ke 28 sebanding dengan penggunaan valaciclovir 2 kali sehari.[39,40] Efek samping utama yang timbul berupa mual, sakit kepala dan muntah.[40]
3. N-methanocarbathymidine (N-MCT, NN-001)N-methanocarbathymidine merupakan nukleosida analog yang memiliki struktur yang terkonformasi tetap dan menunjukkan aktivitas melawan virus herpes dan orthopoxviruse yang lebih efektif dibanding acyclovir. Aktivitas antivirusnya bergantung pada aktivasi dari enzim TK tipe 1 yang akan efektif terhadap virus herpes simplek sedangkan efektivitas terhadap cowpox dan vaccinia virus bergantung pada enzim TK 2.[39]
b. Novel Non-Nucleosidic
Terdapat 2 obat antivirus yang sedang dikembangkan untuk terapi infeksi VZV dan HSV selain analog nukleosida, yakni amenamevir (ASP2151, M5220) (Gambar 6) dan pritelivir (AIC316, BAY 57-1293). Kedua obat ini menargetkan pada kompleks enzim helicase-primase virus yang diperlukan dalam pelepasan DNA virus. Pritelivir merupakan thiazolyl amide yang menunjukkan efikasi yang lebih baik secara spesifik pada HSV 1 dan HSV 2 dibandingkan dengan golongan NA pada studi preklinik. Pritelivir tetap menunjukkan efektivitas terapi meskipun terapi dimulai terlambat.[35,39]
Amenamevir merupakan turunan oxadiazolephenyl dan menunjukkan potensi antivirus yang efektif terhadap VZV maupun HSV. Amenavir diindikasikan sebagai antivirus untuk terapi herpes zoster.

Gambar 6. Struktur helicase-primase inhibitor untuk terapi pada infeksi VZV.[38]
Kesimpulan
Imunitas yang lemah pada pasien imunodefisiensi dan terapi antivirus jangka panjang menjadi faktor yang dapat meningkatkan resistansi VZV terhadap acyclovir. Pasien dengan imunodefisiensi yang terinfeksi VZV resistan acyclovir akan berpotensi mengalami komplikasi yang berat sehingga diperlukan deteksi yang cepat dan penanganan yang sesuai untuk meningkatkan keberhasilan terapi. Pemeriksaan fenotip dan genotip dapat dilakukan untuk mendeteksi resistansi ini sehingga pasien dapat diterapi dengan tepat. Beberapa pilihan antivirus lain telah tersedia untuk dapat dijadikan pilihan terapi dalam menangani infeksi VZV. Namun, efek samping dan toksisitas yang ditimbulkan dari antivirus tersebut menjadi tantangan tersendiri pada manajeman terapi infeksi VZV yang resistan terhadap acyclovir. Saat ini, sudah dikembangkan berbagai antivirus baik kelompok analog nukleosida maupun non nukleosida untuk dapat memberikan hasil terapi yang optimal bagi pasien dengan imunodefisiensi.
DAFTAR PUSTAKA
Oakley A. Herpes zoster. Dalam: Dermatology made easy. Oakley A, penyunting. Banbury: Scion Publishing; 2017: 345-51.
Gershon AA, Breuer J, Cohen JI, Cohrs RJ, Gershon MD, Gilden D, dkk. Varicella zoster virus infection. Nat Rev Dis Primers. 2015; 1: 1-18.
Gueudry J, Boutolleau D, Gueudin M, Burrel S, Miri A, Bodaghi B, Muraine M. Acyclovir- resistant varicella-zoster virus keratitis in an immunocompetent patient. J Clin Virol. 2013; 58(1): 318-20.
Levin MJ, Schmader KE, Oxman MN. Varicella and herpes zoster. Dalam: Fitzpatrick’s Dermatology. Kang S, Amagai M, Bruckner AN, Enk AH, Margolis DJ, McMichael AJ dkk, penyunting. Edisi ke-9. New York: McGraw-Hill; 2019: 3035-64.
Sondakh CC, Kandou RT, Kapantow. Profil varisela di poliklinik kulit dan kelamin RSUP Prof Dr. RD Kandou Manado periode Januari-Desember 2012. Jurnal e-Clinic (eCl). 2015; 3(1): 1-5.
Wiraguna AAGP, Mittaart AH, Zainuddin AS, Muchtar A, Wiryadi BE, Murtiastutik D, dkk. Epidemiologi di Indonesia. Dalam: Buku panduan herpes zoster di Indonesia 2014. Pusponegoro EH, Nilasari H, Lumintang H, Niode NJ, Daili SF, Djauzi S, penyunting. Jakarta: Badan Penerbit Fakultas Kedokteran Universitas Indonesia; 2014:1–60.
Chandrav D. Antiviral activity and mechanism of action of a novel uracil analog for varicella- zoster virus [disertasi]. New York: State University of New York; 2015: 1-193.
Mullane KM, Nuss C, Ridgeway J, Prichard MN, Hartline CB, Theusch J et al. Brincidofovir treatment of acyclovir-resistant disseminated varicella zoster virus infection in an immunocompromised host. Transpl Infect Dis. 2016; 18(5): 785-90.
Andrei G, Snoeck R. Advances in treatment of varicella zoster virus infections. Adv Pharmacol. 2013; 67: 107-68.
Kennedy PGE, Gershon AA. Clinical features of varicella zoster virus infection. Viruses. 2018; 10(11): 609-20.
Satyaprakash A, Ravanfar P, Trying SK. Dermatologic Virology. Dalam: Sauer’s manual of skin diseases. Hall BJ, Hall JC, penyunting. Edisi ke-10. Philadelpia: Wolter Kluwer Health; 2010: 230-9.
Lozano A, Arora A, Mendoza N, MAdkan V, Trying SK. Viral infection. Dalam: Therapy of skin diseases. Krieg T, Bickers DR, Miyachi Y, penyunting. Berlin: Springer. 2010: 157-63.
Sterling JC. Virus infections. Dalam: Rook’s textbook of dermatology. Burns T, Breathnach S, Cox N, Griffiths C, penyunting. Edisi ke-8. West Sussex: Wiley Blackwell; 2010: 1489- 569.
Cohen JI. The varicella-zoster virus genome. Curr Top Microbiol Immunol. 2010; 342: 1-14.
Presti CL, Curti C, Montana M, Bornet C, Vanelle P. Chickenpox: An update. Med Mal Infect. 2019; 49(1): 1-8.
Zaidi Z, Hussain K, Sudhakaran S. Viral infection. Dalam: Treatment of skin disease: A practical guide. Zaidi Z, Hussain K, Sudhakaran S, penyunting. Cham: Springer International Publishing; 2019: 105-8.
Sauerbrei A. Diagnosis, antiviral therapy, and prophylaxis of varicella-zoster virus infections. Eur J Clin Microbiol Infect Dis. 2016; 35(5): 723–34.
Schmidt-Chanasit J, Sauerbrei A. Evolution and world-wide distribution of varicella-zoster virus clades. Infect Genet Evol. 2011; 11(1): 1-10.
Harigane K, Sumi A, Mise K, Kobayashi N. The role of temperature in reported chickenpox cases from 2000 to 2011 in Japan. Epidemiol Infect. 2015; 143(12): 2666-78.
Gershon AA, Gershon MD. Pathogenesis and current approaches to control of varicella-zoster virus infections. Clin Microbiol Rev. 2013; 26(4): 728-43.
Creed R, Satyaprakash A, Trying SK. Varicella Zoster Virus. Mucocutaneous manifestations of viral diseases. Trying SK, More AY, Lupi O, penyunting. Edisi-ke 2. Essex: Informa Health Care; 2010: 98-122.
Piret J, Boivin G. Antiviral resistance in herpes simplex virus and varicella-zoster virus infections: Diagnosis and management. Curr Opin Infect Dis. 2016; 29(6): 654-62.
Kim SR, Khan F, Tyring SK. Varicella zoster: An update on current treatment options and future perspectives. Expert Opin. Pharmacother. 2014; 15(1): 61-71.
Szenborn L, Kraszewska-Głomba B, Jackowska T, Duszczyk E, Majda-Stanisławska E, Marczyńska M, dkk. Polish consensus guidelines on the use of acyclovir in the treatment and prevention of VZV and HSV infections. J Infect Chemother. 2016; 22(2): 65-71.
Brunnemann AK, Bohn-Wippert K, Zell R, Henke A, Walther M, Braum O, dkk. Drug resistance of clinical varicella-zoster virus strains confirmed by recombinant thymidine kinase expression and by targeted resistance mutagenesis of a cloned wild-type isolate. Antimicrob. Agents Chemother. 2015; 59(5): 2726–34.
Piret J, Drouot E, Boivin G. Antiviral drug resistance in herpesviruses. Dalam: Handbook of antimicrobial resistance. Gotte M, Berghuis A, Matlashewski G, Wainberg M, Sheppard D, penyunting. New York: Springer; 2017: 87–122.
Sauerbrei A, Taut J, Zell R, Wutzler P. Resistance testing of clinical varicella-zoster virus strains. Antiviral Res. 2011; 90(3): 242-7.
Van der Beek MT, Vermont CL, Bredius RG, Marijt EW, Van der Blij-de Brouwer CS, Kroes AC, dkk. Persistence and antiviral resistance of varicella zoster virus in hematological patients. Clin Infect Dis. 2013; 56(3): 335-43.
Burrel S, Bonnafous P, Hubacek P, Agut H, Boutolleau. Impact of novel mutations of herpes simplex virus 1 and 2 thymidine kinases on acyclovir phosphorylation activity. Antiviral Res. 2012; 96(3): 386–90.
Malartre N, Boulieu R, Falah N, Cortay JC, Lina B, Morfin F, Frobert E. Effects of mutations on herpes simplex virus 1 thymidine kinase functionality: an in vitro assay based on detection of monophosphate forms of acyclovir and thymidine using HPLC/DAD. Antiviral Res. 2012; 95(3): 224–8.
Brink AATP, Gelder MV, Wolffs PF, Bruggeman CA, Loo IHMV. Compartmentalization of acyclovir resistant varicella zoster virus: Implications for sampling in molecular diagnostics. Clin Infect Dis. 2011; 52(8): 982-7.
Bleymehl K, Cinatl J, Schmidt-Chanasit J. Erratum to: Phenotypic and genetic characterization of varicella-zoster virus mutants resistant to acyclovir, brivudine and/or foscarnet. Med Microbiol Immunol. 2011; 200(3): 193-202.
Perrier M, Désiré N, Deback C, Agut H, Boutolleau D, Burrel S. Complementary assays for monitoring susceptibility of varicella-zoster virus resistance to antivirals. J Virol Methods. 2016; 233: 10-4.
Gnann Jr. JW. Antiviral therapy of varicella-zoster virus infections. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. Chapter 65. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47401/.
Andrei G, Snoeck R. Emerging drugs for varicella-zoster virus infections. Expert Opin Emerging Drugs. 2011; 16(3): 507-35.
Andrei G, Snoeck R. Emerging drugs for varicella-zoster virus infections. Expert Opin Emerging Drugs. 2011; 16(3): 507-35.
Andrei G, Snoeck R. Cidofovir Activity against poxvirus infections. Viruses. 2010; 2(12): 2803-30.
Garikapati S, Nguyen M. Foscarnet. StatPearls Treasure Island. Florida: StatPearls Publishing; 2021: 1-12.
Poole CL, James SH. Antiviral therapies of herpesviruses: Current agents and new directions. Clin Ther. 2018; 40(8): 1282-98.
Birkmann A, Zimmermann H. Drugs in development for herpes simplex and varicella zoster virus. Clin Pharmacol Ther. 2017; 102(1): 30-2.
De SK, Hart JC, Breuer J. Herpes simplex virus and varicella zoster virus: Recent advances in therapy. Curr Opin Infect Dis. 2015; 28(6): 589-95.


